
Heartware Ventricular Assist Device
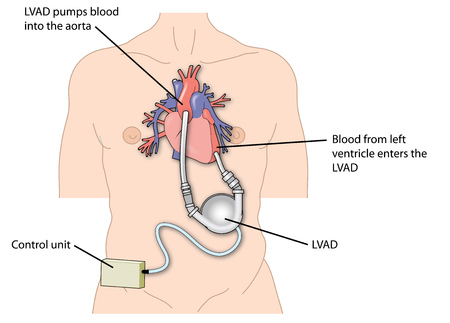
The HeartWare Ventricular Assist Device (HVAD) is used to facilitate blood flow between the heart and the rest of the body in patients waiting for a heart transplant.
How Does the Heartware Ventricular Assist Device Work?
The Heartware Ventricular Assist Device is used in patients awaiting a heart transplant, and who are at risk of death from end-stage left ventricular heart failure. The system can be used in or out of hospital settings, including during patient transport. The apparatus consists of a pump that is surgically implanted in the pericardium, as well a a driveline cable that goes through the skin. The driveline connects the pump to an external controller that regulates the speed and function of the pump. The system is powered with batteries or power adapters.
Because of its small size, the HVAD pump can be implanted through a small incision compared to what was required with older technologies. A physician programs the pump to provide an appropriate amount of blood flow to meet an individual patient’s needs. The Heartware Ventricular Assist Device is designed to pump enough blood every minute to decrease heart failure symptoms.
Medtronic Recalls Heartware Ventricular Assist Device Controllers & DC Adapters
In October 2016, Medtronic announced a recall for HeartWare Controllers due to a power connector problem that could cause the rear portion of the pump’s driveline connector to become separated from the front portion. The recall was later expanded to include HVAD DC Adapters, and now encompasses:
- Serial Numbers: All HeartWare Controllers
- Product Codes: 1400, 1401, 1403, 1407 and 1435 for the HVAD DC Adapter
- Manufacturing Dates: September 30, 2014 to February 29, 2016
A loose connector may allow moisture to enter the HVAD controller, resulting in corrosion, electrical issues, reduced speaker volume and connection failures. A loss of connection could also cause the pump to stop, resulting in death or other serious adverse health consequences. The U.S. Food & Drug Administration (FDA) has designated this action a Class I recall, which indicates that use of the affected products could cause serious or fatal health consequences.
Heartware Splice Kit Recall
In March 2017, Medtronic, Inc. recalled certain Heartware Splice Kits designed to repair the driveline on the Ventricular Assist Device should an electrical break occur. The recall was necessitated by a design problem that would prevent the repaired cable assembly from withstanding excessive force or pull. Such an occurrence could cause damage to the cable assembly and interrupt electrical connection, leading to serious adverse events or death. The FDA designated Medtronic’s action a Class I recall in May 2017.
Lots involved in the recall include those used prior to April 2015 with product codes SY00116 and ASY00281. The recalled Splice Kits were distributed from January 11, 2011 to May 23, 2014.
- FDA (2016) “HeartWare Ventricular Assist Device (HVAD) Pumps by HeartWare Inc.: Class I Recall – Loose Connectors May Prevent Alarm from Sounding” https://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm527148.htm
- FDA (2017) Ventricular Assist Device Controllers and DC Adapter by Medtronic Mechanical Circulatory Support (formerly HeartWare Inc.): Class I Recall – Updated Controller, Power Management Software https://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm556762.htm
- FDA ( 2017) “Medtronic Recalls HeartWare Splice Kit Intended to Repair the Driveline of its Ventricular Assist Device Because it May Cause Electrical Issues or Pump Stop” https://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm527148.htm

Get the latest news and litigation updates about this case by following us on Facebook. Click the "Like" button below.
Follow Us
![]()
![]()
![]()
![]()
![]()
![]()